
La bioinformática como herramienta para el descubrimiento de fármacos
Elena Stephanie Castro Silva y Norma Angélica Estrada Muñoz
En los inicios de la evolución de las supercomputadoras y casi al mismo tiempo que el desarrollo de los videojuegos, la bioinformática surge en los años 60 por la necesidad de interpretar y correlacionar una gran cantidad de datos e información contenida en las secuencias de ADN, ARN y de proteínas. Desde entonces se han creado múltiples herramientas y recursos que prácticamente la han convertido en una disciplina esencial de las ciencias biomédicas y la biotecnología. Contribuye no sólo en la catalogación y caracterización de moléculas de interés farmacológico, sino también en la descripción de las interacciones proteína-proteína, proteína-sustrato/s (o ligando/s), la dinámica a nivel atómico, molecular y subcelular, entre otras aplicaciones [5].
Si quisiéramos visualizar de manera práctica la interacción y/o unión de drogas terapéuticas con un ‘blanco’ específico a nivel molecular —aunque esto ocurre en milisegundos y un poco al azar— podríamos explicarlo como dos piezas de lego que embonan perfectamente o bien, puede que su unión sea interferida por otra pieza o agente distinto [Fig. 1].
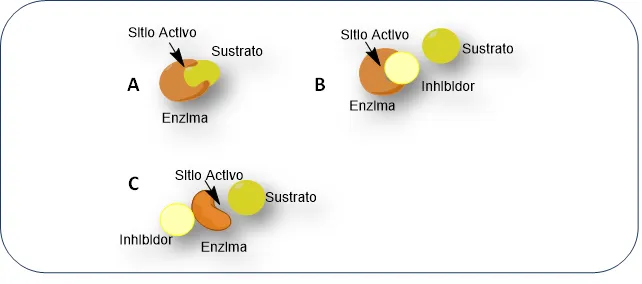
Figura 1.
Los sustratos son moléculas —por ejemplo, metabolitos celulares, hormonas, péptidos o medicamentos sintéticos— que interaccionan con proteínas (enzimas, receptores, anticuerpos), de las que dependen la activación o inhibición de algún proceso biológico, una vez que ambas moléculas coinciden. Sin embargo, aunque pudiera parecer sencillo, la representación y simulación (esquemática, dinámica y/o tridimensional) de las posibles interacciones entre las regiones moleculares clave deben de conocerse a fondo químicamente. Las simulaciones computacionales predicen lo que podría suceder entre las estructuras de dichas moléculas y se basan en características físicas y químicas deseables para que logren interactuar entre sí. A la fecha, se han desarrollado numerosos modelos (de unión entre átomos, grupos químicos y regiones moleculares), cuyos resultados deben ser corroborados adecuadamente en otros sistemas experimentales; en un cierto número de ensayos posteriores se han logrado identificar inhibidores (o activadores) moleculares nuevos, que posiblemente puedan ensayarse y validarse para usarse como fármacos, en la prevención o terapia de ciertos padecimientos [6].
Nuestro caso ilustrativo, en la Fig. 2, muestra un esquema comparativo del acoplamiento molecular en sitios de unión al interior de una enzima, conocida como acetil·colinesterasa humana. Los inhibidores son el fucosterol obtenido del alga parda Sargassum horridum y la neostigmina (un inhibidor comercial); un análisis completo permitió evidenciar que algunos productos naturales marinos (como el fucosterol) pueden ser una fuente de sustancias que inhiban esta enzima, fuertemente asociada con el desarrollo de la enfermedad del Alzheimer [3].
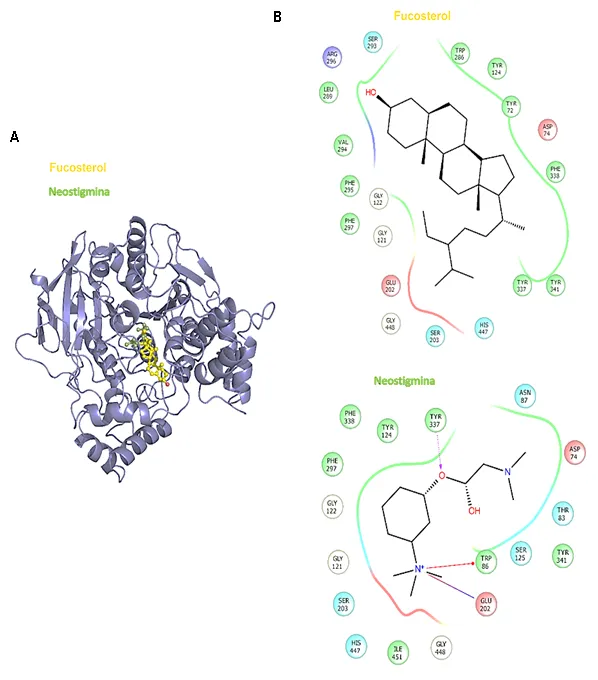
Figura 2.
Simulando como interaccionan las moléculas con potencial terapéutico
Para este tipo de análisis —un procedimiento de ‘acoplamiento molecular’ simulado (docking en inglés)— fue necesario, tanto determinar adecuadamente la estructura química del fucosterol, para observar los aminoácidos de la cadena proteica (en el sitio de unión), con los cuales interacciona esta molécula [Fig. 2], así como corroborar su actividad en ensayos de laboratorio. El programa de análisis Autodock se escogió en base a la accesibilidad, reproducibilidad y sobre todo citación en otros trabajos lo que permitió comparar resultados con otras publicaciones [1, 4, 6].
Es importante señalar que la eficiencia, precisión y velocidad de los resultados obtenidos por estos métodos bioinformáticos dependen en gran medida de varios aspectos: 1) la disponibilidad de tiempo computacional; es decir, si son equipos de uso común o están destinados a desarrollar sólo estos tipos de análisis, así como su capacidad de procesamiento. 2) La plataforma computacional disponible, que se refiere a los programas de análisis de acoplamiento a usar, ya que en la actualidad hay muchos paquetes informáticos de acceso libre.
Algunos de ellos son Autodock, Autodock vina, GOLD (Protein-Ligand Docking Software, por sus siglas en ingles), Glide y Molegro Virtual Docker por mencionar algunos. Así también, los algoritmos matemáticos usados por estos programas y sus cálculos varían según sus funciones, la afinidad con que se une las proteínas y/o su comportamiento y el tipo de inhibidor que se considera generalmente cómo una prioridad en la evaluación del “mejor candidato”. Actualmente, los algoritmos de acoplamiento ya consideran aspectos basados en estructuras químicas completas o solo fragmentos, la ‘flexibilidad’ de las estructuras químicas, las moléculas de agua presentes, la solubilidad y el pH (acidez/ alcalinidad), que podrían influenciar a las estructuras y sus interacciones.
Otro aspecto importante para poder desarrollar un análisis de acoplamiento es si la secuencia de las proteínas de interés está descrita estructuralmente, puesto que una estructura proteica inexacta puede producir un análisis deficiente, mostrando falsas energías de unión, cálculos de interacción/ inhibición erróneos o inexistentes. Por lo tanto, se deben buscar las estructuras químicas más adecuadas al objetivo de la investigación a realizar; en el caso de las proteínas, éstas están accesibles en el [Protein Data Bank](https://www.rcsb.org/) donde se cuentan a la fecha aproximadamente 194,550 registros (secuencias, modelos y otras anotaciones), que permiten avances en la investigación, desarrollo y educación en este ámbito.
Como referencia del aumento en el volumen de trabajos en esta área, en el portal de consulta [PubMed\ del National Council of Biological Information](https://pubmed.ncbi.nlm.nih.gov/?term=molecular docking&timeline=expanded) (NCBI) de los EEUU, encontramos que tan solo para el 2021, existen casi 10 mil 500 publicaciones referidas. Considerando lo anterior, se hace patente que el análisis de acoplamiento molecular es crucial en los requerimientos de los científicos en instituciones de investigación y claramente en la industria farmacéutica, para buscar y evaluar, de manera práctica, rápida y confiable, nuevos compuestos de origen natural o sustancias sintéticas (obtenidas en laboratorio por diversos métodos), que posean características viables como futuros fármacos o medicamentos [2].
Asimismo, todo el proceso fomenta la interdisciplinariedad ya que cualquier análisis requiere de una comprobación objetiva bajo técnicas experimentales; es decir, se deben realizar experimentos de tipo biofísicos y/o bioquímicos que confirmen la unión y la actividad biológica predicha con el acoplamiento. Aunque algunos aspectos de la simulación y predicción requieren aún mejoras tentativas, este tipo de análisis computacionales proporcionan una guía valiosa en facilitar la búsqueda de nuevos fármacos, idealmente, más efectivos, más seguros y accesibles.
Referencias
1. Bello M, M Martinez-Archundia M, & J Correa-Basurto (2013). Automated docking for novel drug Discovery. Expert Opin Drug Discov. 8 (7): 821-834 DOI: 10.1517/17460441.2013.794780
2. Borhani WD & ED Shaw (2012). The future of molecular dynamics simulations in drug discovery J Computer-Aided Molec Des 26:15–26 p. DOI: 10.1007/s10822-011-9517-y
3. Castro-Silva ES, M Bello, M Hernández-Rodríguez, J Correa-Basurto, JI Murillo-Álvarez, MC Rosales-Hernández & M Muñoz-Ochoa (2019). In vitro and in silico evaluation of fucosterol from Sargassum horridum as potential human acetylcholinesterase inhibitor. J Biomol Struct Dyn 37(12): 3259-3268 DOI: [10.1080/07391102.2018.1505551](https://doi.org/10.1080/07391102.2018.1505551)
4. Khamis MA, W Gomaa & WF Ahmed (2015). Machine learning in computational docking. Artif Intell Med 63 (3):135–152 DOI: 10.1016/j.artmed.2015.02.002
5. Ou-Yang SS, JY Lu, XG Kong et al. (2012). Computational Drug Discovery. Acta Pharmacol Sin 33: 1131–1140 DOI: 10.1038/aps.2012.109
6. Prieto-Martínez FD, M Arciniega & JL Medina-Franco (2018). Molecular docking: current advances and challenges. TIP Revista Especializada en Ciencias Químico-Biológicas. Vol. 21(Supl. 1): 65-87. DOI: 10.22201/fesz.23958723e.2018.0.143 (FES-Zaragoza. UNAM).
Comparte este artículo en redes sociales

Acerca de los autores
*La Dra. Castro-Silva realiza actualmente una estancia posdoctoral en colaboración con la Dra. Estrada-Muñoz, del Programa de Acuicultura, en el Centro de Investigaciones Biológicas del Noroeste, S.C. (CIBNOR), en La Paz, B.C.S., México.
Contacto: elenastephaniecastro@gmail.com


