

El papel de la bioinformática en la lucha contra los virus
Blanca Itzelt Taboada Ramírez y Selene Zarate
La epidemiología es la rama de la salud pública que estudia cómo ocurren las enfermedades en una población. Uno de sus objetivos más importantes es el seguimiento de brotes de enfermedades infecciosas. Tradicionalmente, esto se hace registrando un aumento en el número de casos de una enfermedad. Una vez confirmado el brote, se procede a determinar las causas y a rastrear los casos mediante los metodos de diagnóstico disponibles. Dependiendo de que tan caracterizada este la enfermedad, estos métodos pueden identificar subtipos específicos del patógeno o simplemente detectar su presencia.
En el caso de las enfermedades causadas por virus, el avance de las técnicas moleculares ha permitido no solo detectar su presencia, sino también identificar con precisión qué tipos y subtipos circulan en cada región geográfica. Asimismo, la posibilidad de secuenciar genomas virales, es decir, descifrar la totalidad de su material genómico, revolucionó el estudio de las epidemias, permitiendo rastrear su evolución y origen con mayor detalle. El primer virus analizado de esta manera fue el VIH, cuyo análisis genómico en los años 90 reveló su origen zoonótico en chimpancés de la selva del Camerún. La importancia de la pandemia del VIH/SIDA y del virus de influenza promovió el uso de secuenciación genómica para trazar eventos epidémicos, dando origen a la vigilancia genómica.
En la última década, la secuenciación masiva y la bioinformática han cambiado la forma en que respondemos a las epidemias virales. Gracias a estas dos disciplinas, la comunidad científica ahora puede analizar en tiempo real la evolución de los virus, lo que permite a los sistemas de salud a actuar con mayor rapidez y precisión. En este artículo, explicaremos cómo estas herramientas han sido clave en el control de epidemias recientes y su impacto en la salud pública global.
¿Qué es la bioinformática y cómo nos ayuda en la lucha contra las epidemias?
La bioinformática es una ciencia que combina la biología y la computación para analizar datos genómicos y epidemiológicos, como los de un virus. Esto nos ayuda a entender el comportamiento y las características de los virus como el SARS-CoV-2, causante de la COVID-19, o el virus de la Influenza. Estos análisis han permitido describir sus patrones de circulación mundial, y ayudar en la toma de decisiones sobre la actualización de las vacunas para cada temporada. En lugar de esperar meses o años para entender cómo se comporta un virus, la bioinformática nos proporciona acceso inmediato a información crucial como: su estructura genética, sus mutaciones y modo de transmisión entre las personas. Gracias a estas herramientas, podemos contestar preguntas clave como: ¿De dónde viene el virus? ¿Cuánto está cambiando o mutando? ¿Cómo se está transmitiendo en diferentes lugares?
Principales herramientas bioinformáticas para el seguimiento de virus
Para analizar y seguir el rastro de los virus, tenemos varias herramientas que nos ayudan enormemente:
a) Desarrollo de flujos de análisis de datos a partir de experimentos de secuenciación de nueva generación (NGS). Esta tecnología avanzada, de la mano con flujos de trabajo bioinformáticos especializados, permite descifrar el código genético de un virus en horas y es clave para investigar el origen de epidemias, analizando muestras de animales en los que se sospecha que pudo haber surgido un virus, como se muestra en la Figura 1.
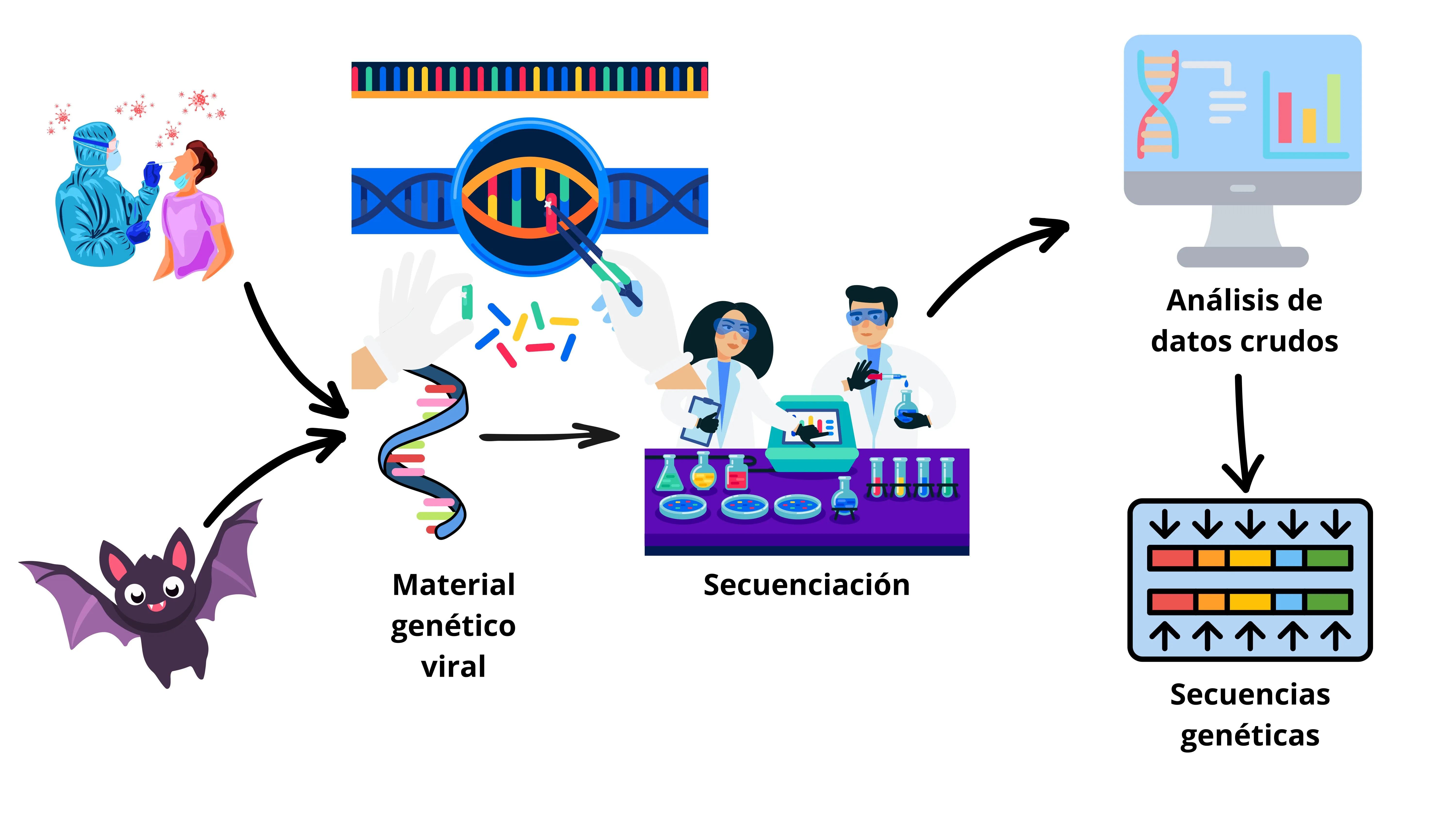
Figura 1. Proceso genómico desde muestras respiratorias o animales: extracción, secuenciación y análisis bioinformático para determinar la secuencia viral.
El proceso inicia con la extracción del material genético del virus a partir de una muestra de una persona o animal infectado, lo que logra mediante técnicas químicas o enzimáticas que rompen las células y partículas virales para liberar su ADN o ARN. Posteriormente, este material genético puede amplificarse de forma específica, de manera que, cuando se introduce en un secuenciador, un dispositivo que lee y determina el orden de los nucleótidos en el ADN o ARN. se genera información principalmente del genoma viral de interés. Estos datos son interpretados con herramientas bioinformáticas con el objetivo de reconstruir la secuencia del genoma viral, revelando su estructura, evolución y mutaciones que pueden afectar su propagación o virulencia.
b) Bases de datos de secuencias virales. Estas herramientas almacenan información genética y facilitan el intercambio de descubrimientos, promoviendo la colaboración global. Plataformas como GISAID y GenBank contienen datos de miles de virus y otros organismos, permitiendo rastrear variantes y patrones de transmisión a nivel mundial.
La Figura 2 muestra el crecimiento de GenBank a lo largo del tiempo, impulsado por tecnologías como la NGS. Este incremento no solo ha fortalecido el estudio de virus, sino que también ha ampliado el conocimiento genético de bacterias, hongos, plantas y otros organismos, contribuyendo a la investigación interdisciplinaria y al manejo de epidemias.
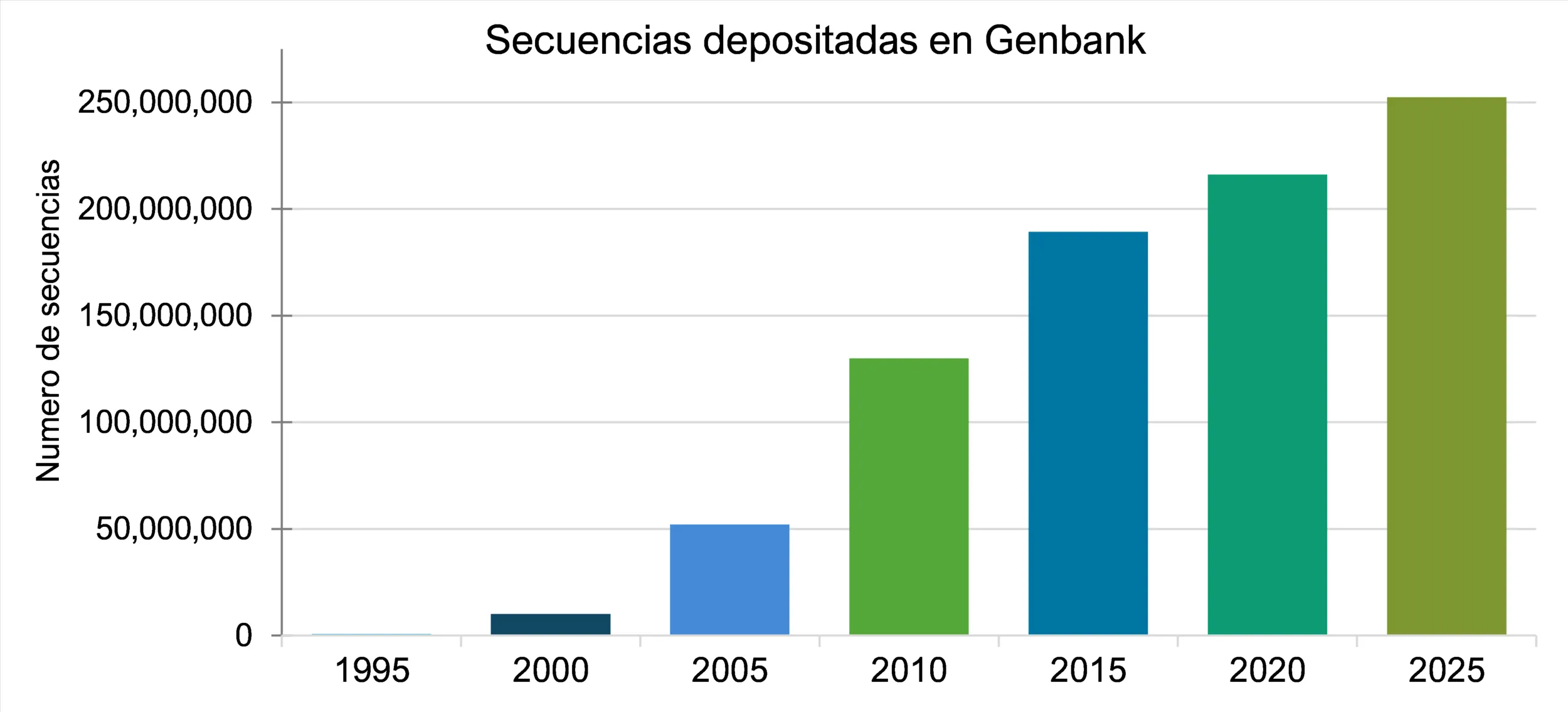
Figura 2. Crecimiento de secuencias en la base de datos de GenBank (1995-2025), impulsado por tecnologías como la Secuenciación de Nueva Generación (NGS).
c) Análisis filogenético. Estas herramientas nos permiten entender cómo están relacionados diferentes virus o variantes de este, basándonos en los cambios genómicos que han acumulado con el tiempo. Son como un árbol genealógico: las ramas representan cómo estamos conectados con nuestros parientes (Figura 3). En los árboles filogenéticos, cuanto más cerca estén dos hojas en el árbol, más reciente es su conexión, como si fueran primos cercanos en una familia. Son especialmente útiles para el seguimiento de enfermedades virales, ya que los virus acumulan cambios genómicos en tiempos comparables a los de los brotes epidémicos, permitiendo un monitoreo casi en tiempo real.
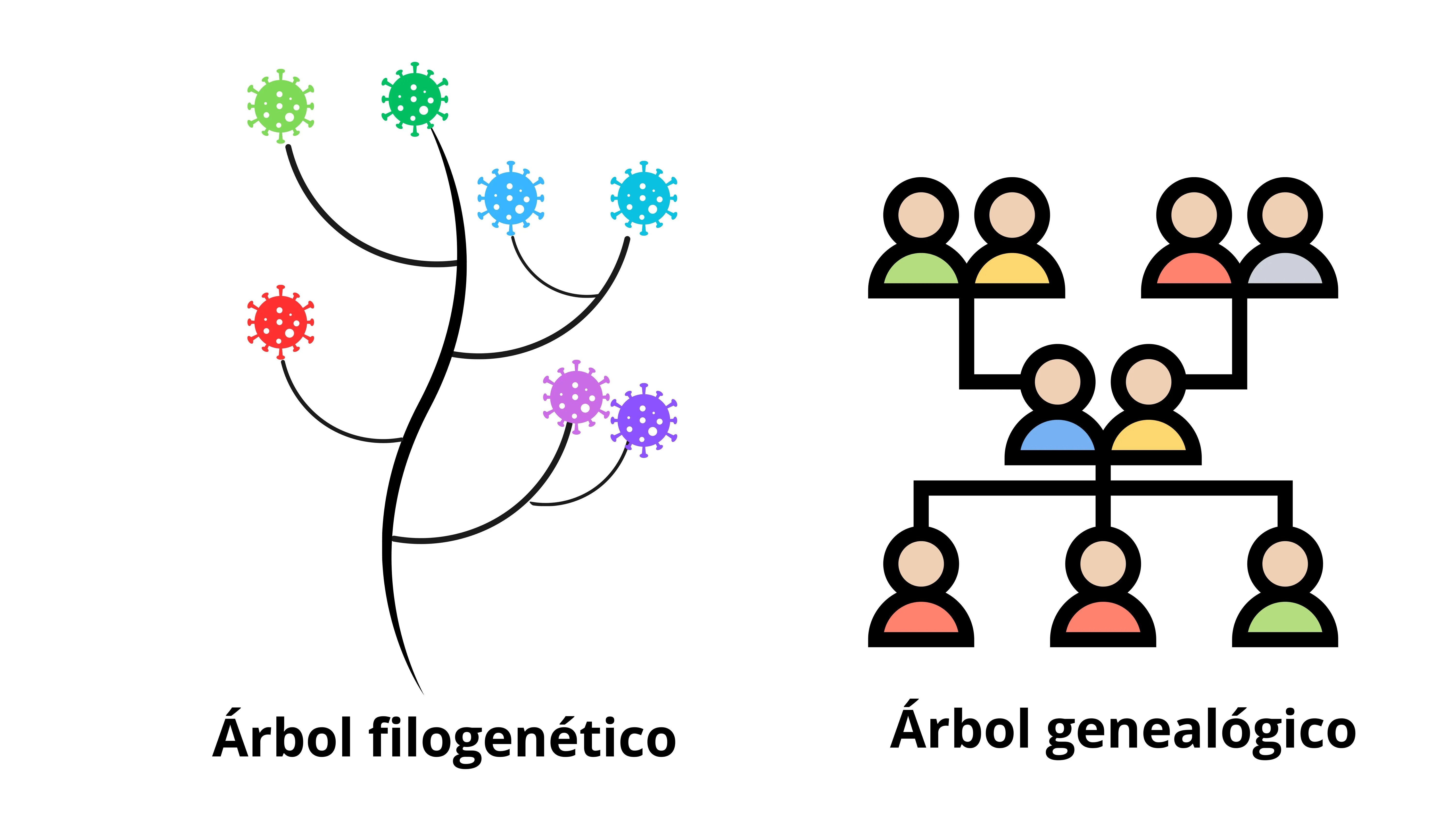
Figura 3. Comparación entre un árbol filogenético y un árbol genealógico. En ambos, la proximidad en la representación gráfica indica una relación ancestro-descendiente más cercana.
d) Modelos de propagación viral. Son herramientas clave que nos ayudan a entender cómo se transmiten los virus y por dónde se desplazan. Estas herramientas combinan información genética, por ejemplo, mutaciones específicas detectadas en las secuencias, con datos espacio-temporales. Esta información permite construir arboles filogenéticos que integran tanto el lugar como la fecha de muestreo de cada secuencia y se conocen como análisis filodinámicos. Gracias a ellos, podemos visualizar su avance en tiempo real y anticipar su comportamiento en diferentes regiones.
Un ejemplo es la herramienta Nextstrain (Figura 4), que analiza datos genómicos del virus junto con información sobre su transmisión, poblaciones afectadas y dinámicas de infección. Esto permite no solo rastrear su recorrido, sino también predecir su expansión. Para ello, emplea modelos matemáticos y estadísticos que integran datos genómicos con información espacio-temporal y epidemiológica, como tasas de infección y movilidad de las poblaciones. Al estudiar la acumulación de mutaciones y su distribución geográfica, es posible inferir rutas de transmisión y estimar la velocidad de propagación del virus, así como proyectar escenarios de dispersión.
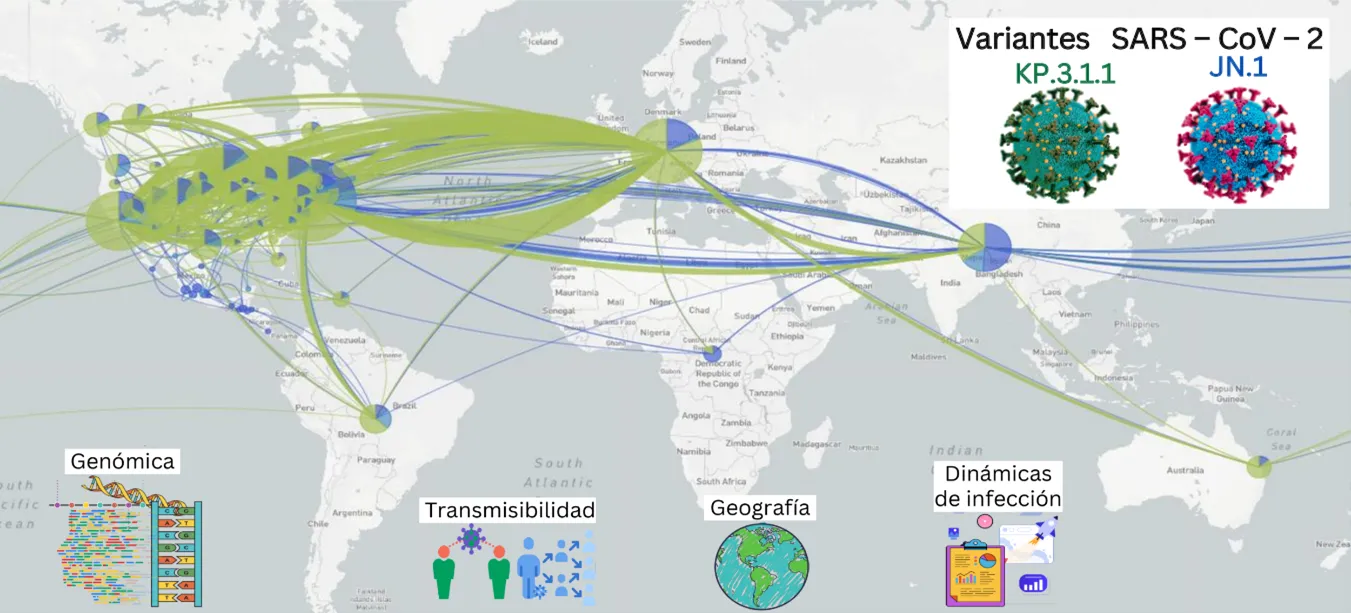
Figura 4. Ejemplo de dispersión y evolución de dos variantes de SARS-CoV-2 analizadas con Nextclade, que integra múltiples factores en su análisis: datos genómicos, que permiten identificar mutaciones clave; patrones de transmisión, que revelan la propagación del virus entre individuos; información geográfica, que muestra la distribución y el flujo de variantes entre países; y datos poblacionales, que ayudan a contextualizar la dinámica de infección.
Estos modelos son esenciales para respaldar decisiones en salud pública. Desde planificar campañas de vacunación hasta establecer restricciones de movilidad, ayudan a proteger a las comunidades y mitigar el impacto de las enfermedades.
Casos de éxito en el uso de herramientas bioinformáticas
La bioinformática ha jugado un papel crucial en varias epidemias recientes, permitiendo respuestas rápidas y efectivas. Durante la pandemia de COVID-19, herramientas como GISAID facilitaron la identificación de variantes del SARS-CoV-2 en México (Figura 5) y a nivel global, mientras que plataformas como Nextstrain visualizaron en tiempo real su evolución y propagación, guiando decisiones clave sobre restricciones a la movilidad y las estrategias de vacunación.
En el brote de Ébola en África Occidental en 2014, la secuenciación de nueva generación y el análisis filogenético facilitaron el rastreo de la transmisión del virus entre comunidades, lo que fue clave para reducir su impacto en áreas vulnerables. Finalmente, el virus de la gripe, que muta constantemente, se ha monitoreado durante años con bases de datos y herramientas bioinformáticas, permitiendo ajustar anualmente la composición de las vacunas para mejorar su eficacia.
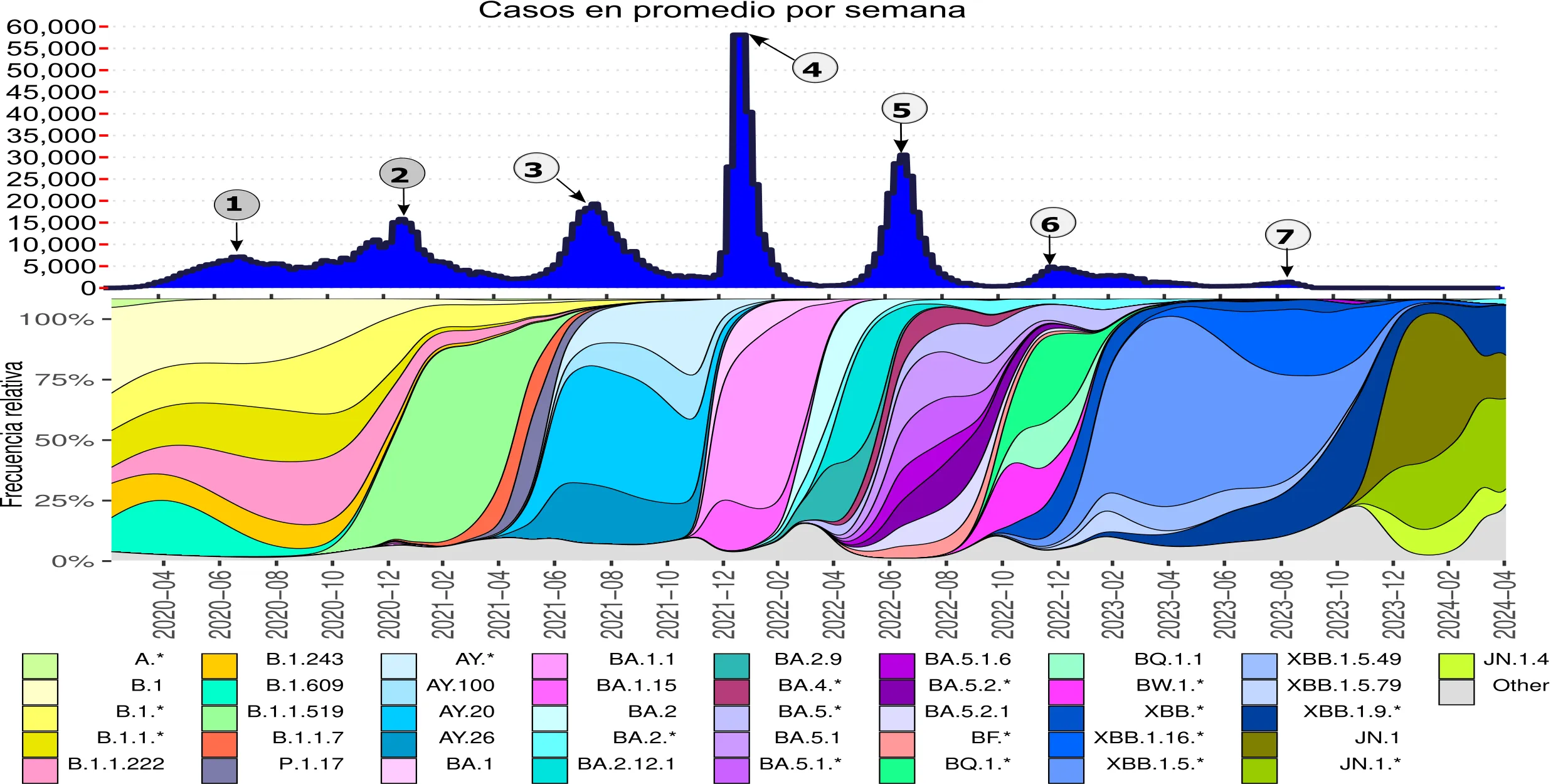
Figura 5. Dinámica de casos promedio semanales y prevalencia de linajes de SARS-CoV-2 en México. La grafica superior muestra los casos semanales promedio, destacando las distintas olas epidemiológicas. La parte inferior presenta la evolución temporal de los linajes virales, donde cada color representa un linaje (identificado por las letras a la derecha de las cajitas de colores en la leyenda). Se observa cómo linajes como B.1.1.519, predominante en la segunda ola, fueron reemplazados por Delta (AY.100, AY.20, AY.26) en la tercera, y posteriormente por Ómicron (BA.1, BA.2, BA.5) en las siguientes olas. En la sexta y séptima olas, sublinajes como XBB.1.5, XBB.1.16 y JN.1 dominaron.
Retos de la bioinformática en el seguimiento de virus y epidemias
Aunque la bioinformática ha avanzado considerablemente, su uso para el seguimiento de epidemias aún enfrenta desafíos que limitan su alcance. Uno de los principales es el acceso desigual a la tecnología, ya que no todas las regiones cuentan con herramientas avanzadas para monitoreo genético en tiempo real. Además, la bioinformática genera enormes volúmenes de datos, lo cual exige computadoras de alta capacidad y personal especializado, recursos que no siempre están al alcance de todos los países.
La bioinformática continúa evolucionando, y en los próximos años veremos varios desarrollos que facilitarán aún más el seguimiento de epidemias, por ejemplo, tecnologías de secuenciación más rápidas y económicas, algoritmos de Inteligencia Artificial que mejorarán los modelos de predicción y nos ayudarán a detectar brotes con mayor rapidez y precisión y una mayor colaboración global, que permitirán respuestas más coordinadas, especialmente ante nuevas amenazas virales.
Lecturas recomendadas
- Cárdenas Guzmán, G. (2014). La vigilancia epidemiológica en México. Revista ¿Cómo ves?. Núm 189. https://www.comoves.unam.mx/numeros/articulo/189/la-vigilancia-epidemiologica-en-mexico
- Ceballos, MA. (2021). ¿Los coronavirus mutantes, me debo preocupar? Revista ¿Cómo ves?. Núm. 266. https://www.comoves.unam.mx/numeros/articulo/268/los-coronavirus-mutantes-me-debo-preocupar
- Duque, H. Evolución viral. Fondo Editorial Biogénesis. 2020, pp. 83–94. https://revistas.udea.edu.co/index.php/biogenesis/article/view/326746.
- Grande, R. Secuenciación masiva del ADN. Biotecnología en Movimiento. 2018. Biotecnología en Movimiento. Número 13. pp. 13-21. https://biotecmov.ibt.unam.mx/services/pdfDownloader.php?id=MTMqKl8qKjQ=
- Zárate, S. Vigilancia genómica de virus en México en la post-pandemia. 2023. Genómicas hoy. Número 25. pp 14.-21.
Comparte este artículo en redes sociales

Acerca de las autoras
La Dra. Blanca ItzetTaboada Ramírez es especialista en ciencias computacionales, con estudios de licenciatura, maestría y doctorado en esta área. Su pasión por la tecnología y la biología la ha llevado a aplicar sus conocimientos en computación al estudio de virus. Forma parte del Consorcio Mexicano de Vigilancia Genómica, donde ha contribuido al seguimiento y análisis de la dispersión y evolución de virus como SARS-CoV-2, Influenza y dengue, entre otros. Además, ha desarrollado herramientas basadas en inteligencia artificial para caracterizar de manera más eficiente estos patógenos, impulsando avances significativos en la comprensión y manejo de enfermedades infecciosas. Actualmente forma parte del Departamento de Genética del Desarrollo y Fisiología Molecular del Instituto de Biotecnología, de la Universidad Nacional Autónoma de México. Selene Zarate realizó sus estudios de licenciatura y doctorado en la UNAM, y dos estancias postdoctorales especializándose en el campo de la evolución de virus. Sus investigaciones se enfocan en estudiar la diversidad y evolución de los virus y cómo éstas se relacionan con la aparición de enfermedades emergentes y el surgimiento de epidemias. Es miembro del Consorcio Mexicano de Vigilancia Genómica creado con el objetivo de contribuir a la vigilancia del virus SARS-CoV-2 durante la pandemia. Además, considera crucial hacer tareas de divulgación de temas relacionados con los virus y los retos que representan actualmente. Forma parte del Posgrado en Ciencias Genómicas de la Universidad Autónoma de la Ciudad de México.
Contacto: blanca.taboada@ibt.unam.mx; selene.zarate@uacm.edu.mx